Химическая энциклопедия
"НИТРОСОЕДИНEНИЯ"
Главная - Химическая энциклопедия - буква Н - НИТРОСОЕДИНEНИЯ
НИТРОСОЕДИНEНИЯ
(С-нитросоединения),
содержат в молекуле одну или неск. нитрогрупп, непосредственно связанных с атомом
углерода. Известны также N- и О-нитро-соединения (см. Нитрамины
и Нитраты органические
). Нитрогруппа имеет строение,
промежуточное между двумя предельными резонансными структурами: Группа планарна; атомы
N и О имеют , sр2-гибридизацию, связи N—О равноценные и практически
полуторные; длины связей, напр. для CH3NO2, 0,122 нм (N—О),
0,147 нм (С—N), угол ONO 127°. Система С—NO2 плоская с низким
барьером вращения вокруг связи С—N. Н., имеющие хотя бы один
а-Н-атом, могут существовать в двух таутомерных формах с общим мезомерным анионом.
О-форма наз. аци
-H. или нитроновой к-той: Известны разл. производные
нитроновых к-т: соли ф-лы RRC=N(O)O- M+ (соли Н.), эфиры
(нитроновые эфиры) и т.д. Эфиры нитроновых к-т существуют в виде иис- и
транс
-изомеров. Существуют циклич. эфиры, напр. N-оксиды изоксазолинов. Назв. Н. производят прибавлением
префикса "нитро" к назв. соединения-основы, по необходимости добавляя
цифровой указатель, напр. 2-нитропропан. Назв. солей Н. производят из назв.
либо С-формы, либо аци
-формы, или нитроновой к-ты. Физические свойства.
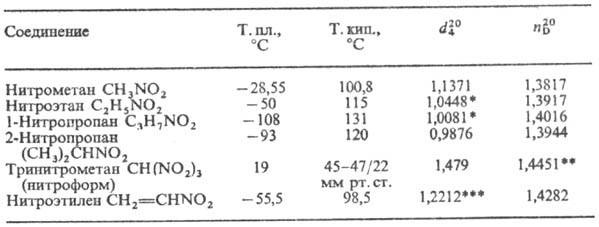
Простейшие нитроалканы-бесцв. жидкости. Физ. св-ва нек-рых алифатических
Н. приведены в таблице. Ароматические Н.-бесцв. или светло-желтые высококипящие
жидкости или низкоплавкие твердые в-ва, обладающие характерным запахом, плохо
раств. в воде, как правило, перегоняются с паром. ФИЗИЧЕСКИЕ СВОЙСТВА
НЕКОТОРЫХ АЛИФАТИЧЕСКИХ НИТРОСОЕДИНЕНИЙ * При 25°С. ** При
24°С. *** При 14°С. В УФ спектрах алифатические
Н. lмакс 200-210 нм (интенсивная полоса) и 270-280 нм (слабая полоса);
для солей и эфиров нитроновых к-т соотв. 220-230 и 310-320 нм; для гем
-динитросоед.
320-380 нм; для ароматических Н. 250-300 нм (интенсивность полосы резко снижается
при нарушении копланарности). В спектре ПМР хим. сдвиги
a-Н-атома в зависимости от строения 4-6 м.д. В спектре ЯМР 14N и
15N хим. сдвиг 5 от - 50 до + 20 м.д. В масс-спектрах алифатических
Н. (за исключением CH3NO2) пик мол. иона отсутствует или
очень невелик; осн. процесс фрагментации - отщепление NO2 или двух
атомов кислорода с образованием фрагмента, эквивалентного нитрилу. Для ароматических
Н. характерно присутствие пика мол. иона; осн. пик в спектре соответствует иону,
получаемому при отщеплении NO2. Химические свойства.
Нитрогруппа - одна из наиб. сильных электроноакцепторных групп и способна
эффективно делокализовать отрицат. заряд. В ароматич. соед. в результате индукционного
и особенно мезомерного эффектов она влияет на распределение электронной плотности:
ядро приобретает частичный положит. заряд, к-рый локализован гл. обр. в орто-
и пара
-положениях; константы Гаммета для группы NO2 sм
0,71, sn 0,778, s+n 0,740, s-n
1,25. Т. обр., введение группы NO2 резко увеличивает реакц. способность
орг. соед. по отношению к нуклеоф. реагентам и затрудняет р-ции с электроф.
реагентами. Это определяет широкое применение Н. в орг. синтезе: группу NO2
вводят в нужное положение молекулы орг. соед., осуществляют разл. р-ции, связанные,
как правило, с изменением углеродного скелета, и затем трансформируют в др.
ф-цию или удаляют. В ароматич. ряду часто используют и более короткую схему:
нитрование-трансформация группы NO2. Мн. превращения алифатических
Н. проходят с предварит. изомеризацией в нитроновые к-ты или образованием соответствующего
аниона. В р-рах равновесие обычно практически полностью сдвинуто в сторону С-формы;
при 20 °С доля аци
-формы для нитрометана 1•10-7, для нитропропана
3.10-3. Нитроновые к-ты в своб. виде, как правило, неустойчивы;
их получают осторожным подкислением солей Н. В отличие от Н. они проводят ток
в р-рах и дают красное окрашивание с FeCl3. Аци-
Н.-более
сильные СН-кислоты (рКа ~ 3-5), чем соответствующие Н. (рКа
~ 8-10); кислотность Н. повышается с введением электроноакцепторных заместителей
в a-положение к группе NO2. Образование нитроновых
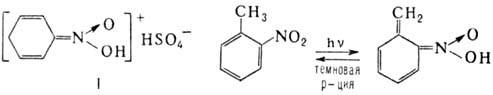
к-т в ряду ароматических Н. связано с изомеризацией бензольного кольца в хиноидную
форму; напр., нитробензол образует с конц. H2SO4 окрашенный
солеобразный продукт ф-лы I, о-нитротолуол проявляет фотохромизм в результате
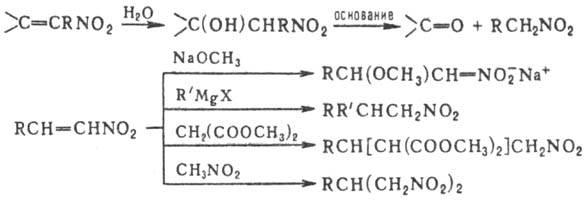
внутримол. переноса протона с образованием ярко-синего О-производного: При действии оснований
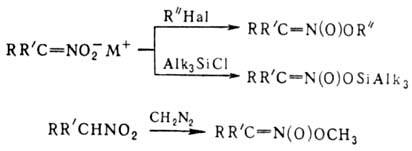
на первичные и вторичные Н. образуются соли Н.; амбидентные анионы солей в р-циях
с электрофилами способны
давать как О-, так и С-производ-ные. Так, при алкилировании солей Н. алкилгалогенидами,
триалкилхлорсиланами или R3O+BF-4
образуются продукты О-алкилирования. Последние м.б. получены также при действии
диазометана либо N,О-бис-(триметилсилил)аце-тамида на нитроалканы с рКа
< 3 или нитроновые к-ты, напр.: Ациклич. алкиловые эфиры
нитроновых к-т термически нестабильны и распадаются по внутримол. механизму: р-цию можно использовать
для получения карбонильных соединений. Более стабильны силиловые эфиры. Об образовании
продуктов С-алкилирования см. ниже. Для Н. характерны р-ции
с разрывом связи С—N, по связям N=O, O=N Р-ц и и с р а з р ы в о
м с в я з и С—N. Первичные и вторичные Н. при нагр. с минер. к-тами в присут.
спиртового или водного р-ра щелочи образуют карбонильные соед. (см. Нефа реакция
). Р-ция проходит через промежут. образование нитроновых к-т: В качестве исходных соед.
можно использовать силиловые нитроновые эфиры. Действие сильных к-т на алифатические
Н. может приводить к гидроксамовым к-там, напр.: Метод используют в пром-сти
для синтеза СН3СООН и гидроксиламина из нитроэтана. Ароматические
Н. инертны к действию сильных к-т. При действии восстановителей
(напр., TiCl3-H2O, VCl2-Н2О-ДМФА)
на Н. или окислителей (KMnO4-MgSO4, O3) на
соли Н. образуются кетоны и альдегиды. Алифатические Н., содержащие
подвижный атом Н в b-положении к группе NO2, при действии оснований
легко элиминируют ее в виде HNO2 с образованием олефинов. Аналогично
протекает термич. разложение нитроалканов при т-рах выше 450°. Вицинальные
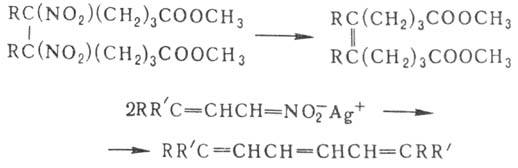
динитросоед. при обработке амальгамой Са в гексамстаноле отщепляют обе группы
NO2, Ag-соли непредельных Н. при потере групп NO2 способны
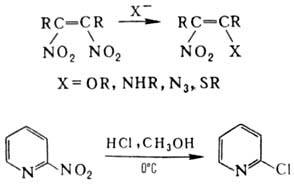
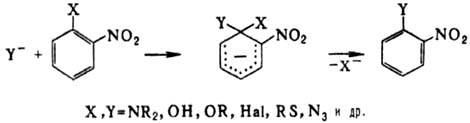
димеризоваться: Нуклеоф. замещение группы
NO2 не характерно для нитроалканов, однако при действии тиолат-ионов
на третичные нитроалканы в апротонных р-рителях группа NO2 замещается
на атом водорода. Р-ция протекает по анион-радикальному механизму. В алифатич.
и гетероциклич. соед. группа
NO2 при кратной связи относительно легко замещается на нуклеофил,
напр.: В ароматич. соед. нуклеоф.
замещение группы NO2 зависит от ее положения по отношению к др. заместителям:
группа NO2, находящаяся в мета
-положении по отношению к электроноакцепторным
заместителям и в орто-
и пара-
положениях к электронодонорным,
обладает низкой реакц. способностью; реакц. способность группы NO2,
находящейся в орто-
и пара
-положениях к электроноакцепторным заместителям,
заметно увеличивается. В нек-рых случаях заместитель вступает в орто
-положение
к уходящей группе NO2 (напр., при нагр. ароматических Н. со спиртовым
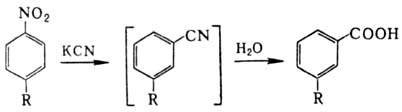
р-ром KCN, р-ция Рихтера): Р-ц и и п о с в я з и N
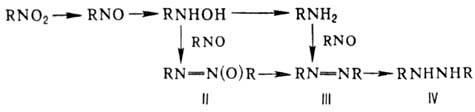
= O. Одна из важнейших р-ций-вос-становление, приводящее в общем случае к набору
продуктов: Азокси-(II), азо-(III)
и гидразосоед. (IV) образуются в щелочной среде в результате конденсации промежуточно
возникающих нитрозосоед. с аминами и гидроксиламинами. Проведение процесса в
кислой среде исключает образование этих в-в. Нитрозосоед. восстанавливаются
быстрее, чем соответствующие Н., и выделить их из реакц. смеси, как правило,
не удается. Алифатические Н. восстанавливаются в азокси- или азосоединения при
действии алкоголятов Na, ароматические-при действии NaBH4, обработка
последних LiAlH4 приводит к азосоединениям. Электрохим. восстановление
ароматических Н. при определенных условиях позволяет получить любое из представленных
производных (за исключением нитрозосоед.); этим же методом удобно получать гидроксиламины
из мононитроалканов и амидоксимы из солей гем
-динитроалканов: Известно много методов
восстановления Н. до аминов. Широко используют железные опилки, Sn и Zn в присут.
к-т; при каталитич. гидрировании в качестве катализаторов используют Ni-Ренея,
Pd/C или Pd/PbCO3 и др. Алифатические Н. легко восстанавливаются
до аминов LiAlH4 и NaBH4 в присут. Pd, амальгамами Na
и Аl, при нагр. с гидразином над Pd/C; для ароматических Н. иногда применяют
ТlСl3, СrСl2 и SnCl2, ароматич. поли-Н. избирательно
восстанавливаются до нитраминов гидросульфидом Na в СН3ОН. Существуют
способы избират. восстановления группы NO2 в полифункциональных Н.
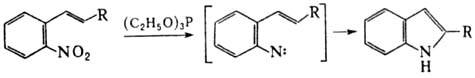
без затрагивания др. ф-ций. При действии Р(III) на
ароматические Н. происходит последоват. дезоксигенирование группы NO2
с образованием высокореакционноспособных нитренов. Р-цию используют для синтеза
конденсир. гетероциклов, напр.: В этих же условиях силиловые
эфиры нитроновых к-т трансформируются в силильные производные оксимов. Обработка
первичных нитроалканов РСl3 в пиридине или NaBH2S приводит
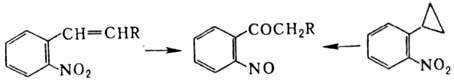
к нитрилам. Ароматические Н., содержащие в орто
-положении заместитель
с двойной связью или циклопропильный заместитель, в кислой среде перегруппировываются
в о-нитрозокетоны, напр.: Н. и нитроновые эфиры реагируют
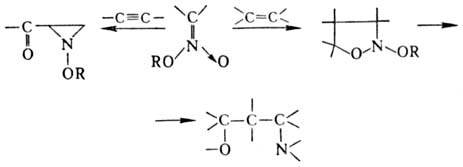
с избытком реактива Гриньяра, давая производные гидроксиламина: Р-ции по связям O = N Наиб. легко эта р-ция протекает
между нитроновыми эфира-ми и олефинами или ацетиленами. В продуктах циклоприсоединения
(моно- и бициклич. диалкоксиаминах) под действием нуклеоф. и электроф. реагентов
связи N — О легко расщепляются, что приводит к разл. алифатич. и гетеро-циклич.
соед.: В препаративных целях в
р-ции используют стабильные силиловые нитроновые эфиры. Р-ц и и с с о х р а н е
н и е м г р у п п ы NO2. Алифатические Н., содержащие a-Н-атом, легко
алкилируются и ацилируются с образованием, как правило, О-производных. Однако
взаи-мод. дилитиевых солей первичных Н. с алкилгалогенидами, ангидридами или
галогенангидридами карбоновых к-т приводит к продуктам С-алкилирования или С-ацилирования,
напр.: Известны примеры внутримол.
С-алкилирования, напр.: Первичные и вторичные Н.
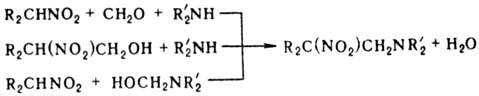
реагируют с алифатич. аминами и СН2О с образованием р-аминопроизводных
(р-ция Манниха); в р-ции можно использовать предварительно полученные метилольные
производные Н. или аминосоед.: Нитрометан и нитроэтан
могут конденсироваться с двумя молекулами метилоламина, а высшие нитроалканы-
только с одной. При определенных соотношениях реагентов р-ция может приводить
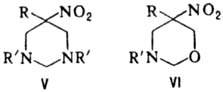
к гетероциклич. соед., напр.: при взаимод. первичного нитроалкана с двумя эквивалентами
первичного амина и избытком формальдегида образуются соед. ф-лы V, если реагенты
берут в соотношении 1:1:3-соед. ф-лы VI.
Ароматические Н. легко
вступают в р-ции нуклеоф. замещения и значительно труднее-в р-ции электроф.
замещения; при этом нуклеофил на правляется в орто-
и пора-поло
жения, а электрофил-в мета-
положение к группе NO2. Константа
скорости электроф. нитрования нитробензола на 5-7 порядков меньше,
чем бензола; при этом образуется м-динитробензол. Активирующее влияние группы
NO2 на нуклеоф. замещение (особенно по орто
-положению) широко
используют в орг. синтезе и пром-сти. Р-ция протекает по схеме присоединение-отщепление
с промежут. образованием s-комплек-са (комплекс Майзенхаймера). По этой схеме
атомы галогенов легко замещаются на нуклеофилы: Известны примеры замещения
по анион-радикальному механизму с захватом электрона ароматич. соединением и
выбросом галогенид-иона или др. групп, напр. алкокси, амино, сульфатной, NO-2.
В последнем случае р-ция проходит тем легче, чем больше отклонение группы NO2
от копланарности, напр.: в 2,3-динитротолуоле замещается в осн. группа NO2
в положении 2. Атом Н в ароматических Н. также способен к нуклеоф. замещению-нитробензол
при нагр. с NaOH образует o-нитрофенол. Нитрогруппа облегчает перегруппировки
ароматич. соед. по механизму внутримол. нуклеоф. замещения или через стадию
образования карбанионов (см. Смайлса перегруп-пировка). Введение второй группы
NO2 ускоряет нуклеоф. замещение. Н.
в присут. оснований присоединяются к альдегидам и кетонам, давая нитроспирты
(см. Анри реакции
), первичные и вторичные Н.-к соед., содержащим активир.
двойную связь (р-ция Михаэля), напр.: Первичные Н. могут вступать
в р-цию Михаэля со второй молекулой непредельного соед.; эту р-цию с послед.
трансформацией группы
NO 2 используют для синтеза поли-функцион. алифатич. соединений.
Комбинация р-ций Анри и Михаэля приводит к 1,3-динитросоединениям, напр.: К неактивир. двойной связи
присоединяются лишь Hg-производные гем-
ди- или тринитросоединений,
а также IC(NO2)3 и C(NO2)4, при
этом образуются продукты С- или О-алкилирования; последние могут вступать в
р-цию цикло-присоединения со второй молекулой олефина: Легко вступают в р-ции
присоединения нитроолефины: с водой в слабокислой или слабощелочной среде с
послед. ретрореакцией Анри они образуют карбонильные соед. и нитроалканы; с
Н., содержащими a-Н-атом,-поли-Н.; присоединяют и др. СН-кислоты, такие, как
ацетилацетон, эфиры ацетоуксусной и малоновой к-т, реактивы Гриньяра, а также
нуклеофилы типа OR-, NR-2 и др., напр.: Нитроолефины могут выступать
в роли диенофилов или диполярофилов в р-циях диенового синтеза и циклоприсое-динения,
а 1,4-динитродиены-в роли диеновых компонентов, напр.: Нитрозирование первичных
Н. приводит к нитроловым к-там RC(=NOH)NO2, вторичные Н. образуют
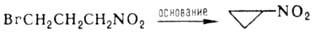
псевдо-нитролы RRC(NO)NO2, третичные Н. в р-цию не вступают. Нитроалканы легко галогенируются
в присут. оснований с последоваг. замещением атомов Н при a-С-атоме: При фотдхйм. хлорировании
замещаются более удаленные атомы Н: При карбоксилировании первичных
нитроалканов действием CH3OMgOCOOCH3 образуются a-нитрокарбоновые
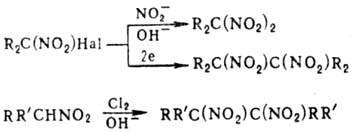
к-ты или их эфиры. При обработке солей моно-Н.
C(NO2)4., нитритами Ag или щелочных металлов либо при
действии нитритов на a-гало-геннитроалканы в щелочной среде (р-ция Тер Меера)
образуются гем
-динитросоединения. Электролиз a-галоген-нитроалканов в
апротонных р-рителях, а также обработка Н. Сl2 в щелочной среде или
электроокисление солей Н. приводят к виц
-динитросоединениям: Нитрогруппа не оказывает
существ. влияния на свободно-радикальное алкилирование или арилирование ароматич.
соед.; р-ция приводит в осн. к орто-
и пара
-замещенным продуктам. Для восстановления Н. без
затрагивания группы NO2 применяют NaBH4, LiAlH4
при низких т-рах или р-р дибора-на в ТГФ, напр.: Ароматич. ди- и три-Н.,
в частности 1,3,5-тринитробен-зол, образуют устойчивые ярко окрашенные кристаллич.
мол. комплексы с ароматич. соед.-донорами электронов (аминами, фенолами и др.).
Комплексы с пикриновой к-той используют для выделения и очистки ароматич. углеводородов.
Взаимод. ди- и тринитробензолов с сильными основаниями (НО-, RO-,
N-3, RSO-2, CN-, алифатич.
аминами) приводит к образованию комплексов Майзен-хаймера, к-рые выделяют в
виде окрашенных солей щелочных металлов. Получение. В пром-сти
низшие нитроалканы получают жидкофазным (р-ция Коновалова) или парофазным (метод
Хэсса) нитрованием смеси этана, пропана и бутана, выделяемых из природного газа
или полученных переработкой нефти (см. Нитрование
). Таким методом получают
и высшие Н., напр. нитроциклогексан - полупродукт в произ-ве капролактама. В лаборатории для получения
нитроалканов применяют нитрование азотной к-той соед. с активир. метиленовой
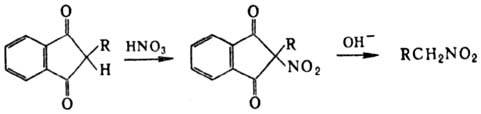
группой; удобный метод синтеза первичных нитроалканов -нитрование 1,3-индандиона
с послед. щелочным гидролизом a-нитрокетона: Алифатические Н. получают
также взаимод. AgNO2 с алкилгалогенидами или NaNO2 с эфирами
a-галогенкарбо-новых к-т (см. Мейера реакция
). Алифатические Н. образуются
при окислении аминов и оксимов; окисление оксимов -способ получения гем
-ди-
и гем
-тринитросоединений, напр.: Нитроалканы м.б. получены
нагреванием ацилнитратов до 200 °С. Мн. методы синтеза Н. базируются
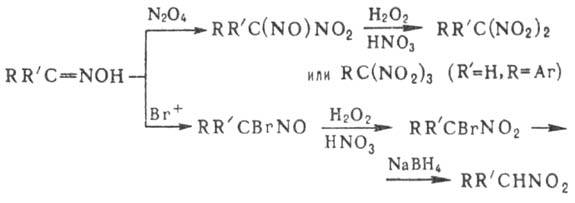
на нитровании олефи-нов оксидами азота, HNO3, солями нитрония, NO2Cl,
орг. нитратами и т.п. Как правило, при этом получают смесь виц
-динитросоединений,
нитронитратов, нитронитритов, непредельных Н., а также продуктов сопряженного
присоединения группы NO 2 и молекулы р-рителя или продуктов их гидролиза,
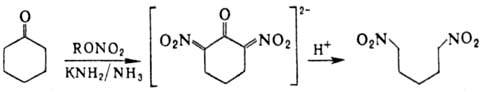
напр.: a,w-Динитроалканы получают
действием алкилнитратов на циклич. кетоны с послед. гидролизом солей a,a-динитро-кетонов: Поли-Н. синтезируют деструктивным
нитрованием разл. орг. соед.; напр., три- и тетранитрометан получают при действии
HNO3 на ацетилен в присут. ионов Hg(II). Осн. метод получения ароматических
Н.-электроф. нитрование. Активная нитрующая группа-ион нитрония NO2,
генерируемый из HNO3 при действии сильных протонных или апротонных
к-т. Для нитрования в мягких условиях используют соли нитрония (NO2BF4,
NO2ClO4 и т.п.), а также N2O5 в
инертных р-рителях. В пром-сти для нитрования
ароматич. соед. используют, как правило, нитрующие смеси (H2SO4
+ HNO3). В лаборатории для повышения концентрации иона нитрония вместо
H2SO4 применяют АlСl3, SiCl4, BF3
и т.п., часто нитрование проводят в инертных р-рителях (СН3СООН,
сульфолан, нитрометан и т.п.). Легко заменяются на группу NO2 сульфо-
и диазогруппы. Для введения в нитробензол второй группы NO 2 в орто-
и пара
-положения вначале получают соответствующее диазопроизводное,
а затем замещают диазогруппу по р-ции Зандмейера. Ароматические Н. получают
также окислением нитрозо-, диазо- и аминогрупп. Применение. Поли-Н.,
особенно ароматические, применяют в качестве взрывчатых веществ
и в меньшей
степени как компоненты ракетных топлив. Алифатические Н. используют как р-рители
в лакокрасочной пром-сти и в произ-ве полимеров, в частности эфиров целлюлозы;
для очистки минер. масел; депарафинизации нефти и др. Ряд Н. находят применение
в качестве биологически активных в-в. Так, эфиры фосфорной к-ты, содержащие
нитроарильный фрагмент,-инсектициды; производные 2-нитро-1,3-пропандиола и 2-нитростирола
- фунгициды; производные 2,4-динитрофенола - гербициды; a-нитрофураны -важнейшие
антибактериальные препараты, на их основе созданы лекарства, обладающие широким
спектром действия (фуразолидин и др.). Нек-рые ароматические Н.-душистые в-ва. Н.- полупродукты в произ-ве
синтетич. красителей, полимеров, моющих препаратов и ингибиторов коррозии; смачивающих,
эмульгирующих, диспергирующих и флотац. агентов; пластификаторов и модификаторов
полимеров, пигментов и пр. Они находят широкое применение в орг. синтезе и в
качестве модельных соед. в теоретич. орг. химии. Нитропарафины обладают
сильным местным раздражающим действием и являются относительно токсичными в-вами.
Относятся к клеточным ядам общего действия, особенно опасны для печени. ЛД50
0,25-1,0 г/кг (при пер-оральном введении). Хлорированные и непредельные Н. в
5-10 раз токсичнее. Ароматические Н. угнетают нервную и особенно кровеносную
систему, нарушая снабжение организма кислородом. Признаки отравления - гиперемия,
по-выш. выделение слизи, слезотечение, кашель, головокружение, головная боль.
Ср-ва первой помощи-хинин и кислород. Метаболизм Н. связан с окислит.-восстановит.
р-циями и, в частности, с окислит. фосфорилированием. Напр., 2,4-динитрофенол
- один из наиб. мощных реагентов, разобщающих процессы окисления и фосфорилирования,
что препятствует образованию АТФ в клетке. В мире производится несколько
сотен различных Н. Объем произ-ва важнейших алифатических Н.-десятки тыс. т,
ароматических-сотни тыс. т; напр., в США производится 50 тыс. т/год нитроалканов
С1-С3 и 250 тыс. т/год нитробензола. См. также м-Динитробензол
,
Нитроанизолы
, Нитробензол
, Нитрометап, Нитротолуолы
и др. Лит.: Химия нитро-
и нитрозогрупп, под ред. Г. Фойера, пер. с англ., т. 1-2, М., 1972-73; Химия
алифатических и алициклических нитросоединений, М., 1974; Общая органическая
химия, пер. с англ., т. 3, М., 1982, с. 399-439; Тартаковский В. А., "Изв.
АН СССР. Сер. хим.", 1984, № 1, с. 165-73. В. А. Тартаковский. |

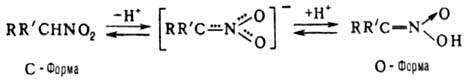
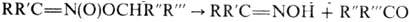 ;
эту
;
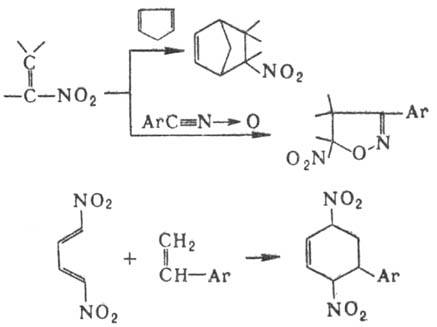
эту О, C=N -> О и р-ции с сохранением группы NO2.
О, C=N -> О и р-ции с сохранением группы NO2.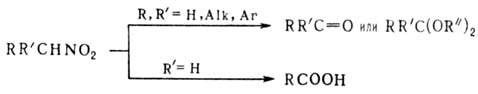
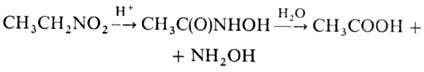
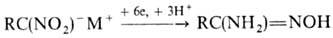
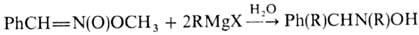
 О и C = N
О и C = N  О. Н. вступают в р-ции 1,3-диполярного циклоприсоединения, напр.:
О. Н. вступают в р-ции 1,3-диполярного циклоприсоединения, напр.: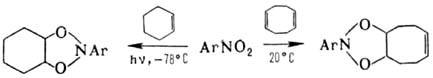
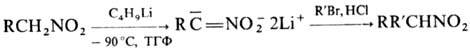
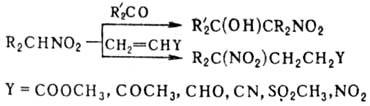
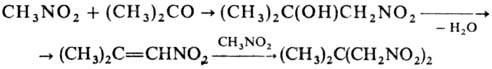
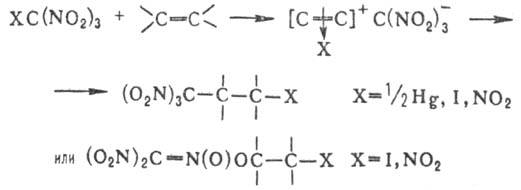
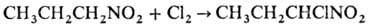
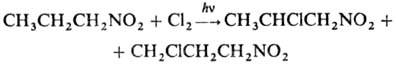
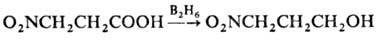
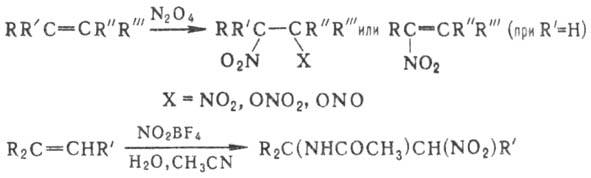
Поделитесь с друзьями:
Вы можете поставить ссылку на это слово:
будет выглядеть так: НИТРОСОЕДИНEНИЯ
будет выглядеть так: Что такое НИТРОСОЕДИНEНИЯ